Directory
- Nomenclature
- Use BLAST
- Browse genome using GBrowse
- Query a single gene
- Search a group of genes
- Query insect orthology
- Browse biological pathways
- Query ESTs and migratory profiles
- Fetch genomic sequence
- Browse expanded and contracted gene families
- Browse microRNAs
- Monarch migration biology
Nomenclature
Use the appropriate formats when searching data with IDs:
| Assembly v3 | DPSCF3xxxxx |
| Assembly v1 | scaffoldxxxxx => DPSCF1xxxxx |
| Geneset OGS2.0 | DPOGS2xxxxx |
| Geneset OGS1.0 | DPGLEANxxxxx => DPOGS1xxxxx |
| KGM_xxxxx => DPOGS1xxxxx | |
| Monarch EST | BF14_xxxx_C1 or BF010xxxxxxx |
| Ortholog group | MCL_xxxxx |
| Gene Ontology | GO:xxxxxxx |
| InterPro domain | IPRxxxxxx |
| KEGG orthology | Kxxxx |
| KEGG pathway | koxxxxx |
Use BLAST
For BLAST, use sequences as input to search for monarch scaffolds, contigs, genes, ESTs, as well as proteins of other insect orders. Select the appropriate search programs and databases. Advanced users can also set the optional parameters to filter the result.
| Databases for BLASTN, TBLASTN, and TBLASTX: |
| Monarch genome Assembly v3: Latest version of assembly |
| Monarch genome Assembly v1: Previous version of assembly |
| Monarch genome scaffolds v0: Initial assembly without filtering |
| Monarch genome contigs v0: Initial contigs before scaffolding |
| Monarch genes OGS2.0 [CDS]: Latest version of geneset |
| Monarch genes OGS1.0 [CDS]: Previous version of geneset |
| Monarch ESTs: expressed sequence tags of monarch brain |
| Databases for BLASTP and BLASTX: |
| Monarch genes OGS2.0 [PEP]: Latest version of geneset |
| Monarch genes OGS1.0 [PEP]: Previous version of geneset |
| Insect proteins: A collection of 332,930 proteins of 20 insect species |
MonarchBase uses html4blast to customize BLAST output. Thus the generated hits in the result page can be linked to additional pages. Genomic sequence is set to link to the GBrowse interface:

Gene sequence is set to link to the gene page:
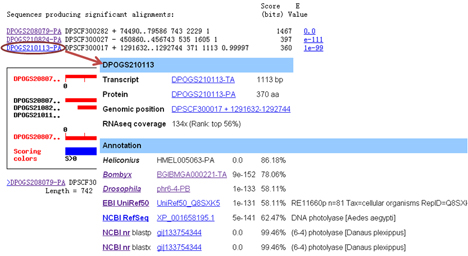
[Use BLAST] [Go back to the menu]
Browse genome using GBrowse
Genome browsers enable users to visualize and browse entire genomes with annotated data.
Through GBrowse of MonarchBase,
the following data can be browsed along with the monarch assembly:
Official geneset:
OGS2.0 is the latest version of the monarch official geneset.
Consensus gene models:
Consensus gene models were generated considering ab initio predicted
genesets, monarch cDNA evidence, and insect homology evidence. The consensus gene models are superior to any independent set
in overall quality. GLEAN
and MAKER are two independent methods.
The GLEAN set was finally adopted as our official geneset, as our quality controls showed that it is
superior to
Maker. The Maker set is also helpful, as it reports entire transcripts; while GLEAN models only include
the CDS region.
Ab initio geneset:
Ab initio programs predict genes based on underlying mathematical models describing
patterns of intron/exon structure and consensus start signals. Because each gene prediction program
currently in use has both strengths and weaknesses, we used five different ab initio methods,
AUGUSTUS,
GeneMark.HMM,
Genscan,
GlimmerHMM,
and SNAP, to generate
preliminary genesets, which were further used as inputs for consensus models. Displaying all prediction
sets is helpful to optimize gene models when there are conflicting overlaps between consensus sets.
Homolog and cDNA evidence:
Aligning monarch cDNA sequences or protein sequences of other insect species helps identify sequence
regions that are likely associated with a gene. Monarch EST alignment indicates the location of
brain-derived ESTs (expressed sequence tags).
The displayed hits are also an entrance site to migratory profiles, which helps
construct the connections between genes and expression data. Monarch RNAseq assembly indicates
the assembled transcripts (by Cufflinks) using
our RNAseq library. This is helpful to show alternative splicing patterns, as well as the untranslated
regions (UTRs)
that were not included in the OGS2.0 gene sequences. GeneWise Bombyx and GeneWise Heliconius
indicate two independent genesets that were generated by GeneWise method using proteins of other two
lepidopteran species. Homologs alignment indicates the TBLASTN hits
of other insect proteins and human proteins.
Features:
Repeat represents monarch repetitive elements that were identified by
RepeatMasker or
repeatrunner. Exercise care
when gene models overlap with a repeat. Some low-complexity repeats can
align to low-complexity protein regions, creating a false sense of homology throughout the genome.
High-complexity repeats often encode real proteins, which are problematic with ab initio predictors. For
example, a transposable element that occurs next to or even within the intron of a protein encoding
gene might mislead predictors to include extra exons as part of a gene model; that is, sequence that does not
belong to the coding sequence of the gene. tRNAs (transfer RNAs) were predicted by
tRNAscan-SE. rRNAs (ribosomal RNAs)
were predicted by RNAmmer and
Rfam scan pipeline.
When you use GBrowse for the first time, select the tracks representing the data type you desire:
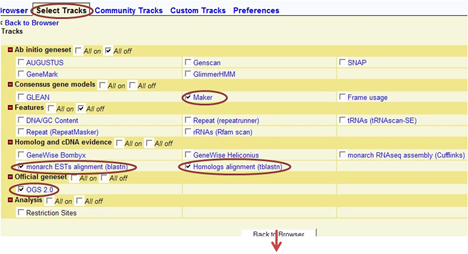
Simply by clicking on the displayed track, you can get access to gene pages, exon sequences, or EST profiles:
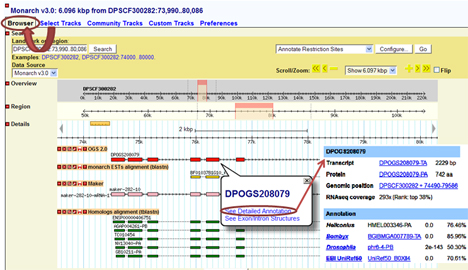
Detailed user tutorial for GBrowse can be found at OpenHelix.
[Browse genome] [Go back to the menu]
Query a single gene
Each OGS2.0 gene has a single gene page. Each monarch gene identifier in all MonarchBase components has been link to its gene page. You can also retrieve gene pages directly by inputing gene ID or keywords:
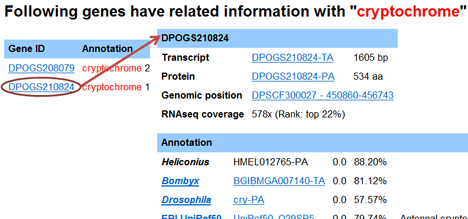
Genomic position shows the name of the scaffold, the strand of gene location, and start and stop positions. You can gain access to the GBrowse interface by clicking on the Genomic position link:
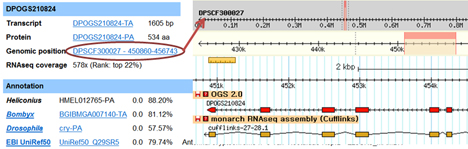
RNAseq coverage is the normalized sequencing depth of our RNAseq library, which represents multiple developmental stages and tissues (Zhan et al. 2011). Rank (from high to low) helps you know the relative level of the expression value.
Annotation was reported according to BLASTP against several insect genesets and public databases. EBI UniRef50 and NCBI RefSeq collect well-annotated proteins, which help report proper annotaion. NCBI nr indicates the non-redundant nucleotide collection, which includes a broader scope of genes. On the other hand, if a gene shows different annotation between BLASTP and BLASTX, it could be considered a potential pseudogene.
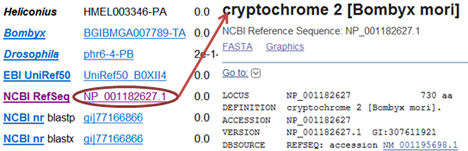
Genes were also assigned to gene families or pathways. Click on the identifiers to access the group page to check detailed information and other related genes:

The Nucleotide sequence and deduced Protein sequence are displayed in FASTA format. Note that untranslated regions (UTRs) were NOT included here. You can retrieve UTRs through GBrowse, according to the gene models of RNAseq (Cufflinks) or EST alignment:

[Query genes] [Go back to the menu]
Search a group of genes
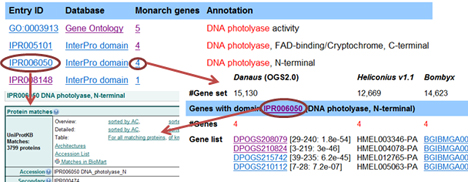
We clustered monarch genes into functional groups or biological pathways. You can search a group of genes for GO term, KO, InterPro domain, or ortholog group. You can use either IDs or keywords as input to search:
You can also search a list of genes having BLAST hits with your input sequence(s). This function is helpful to find all candidate homologs for designated genes:
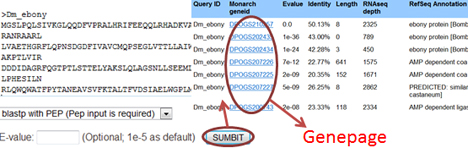
[Query gene families] [Go back to the menu]
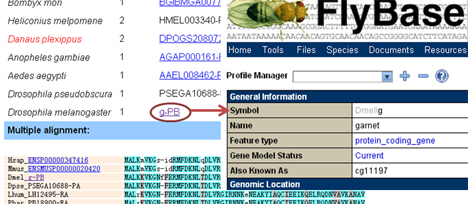
Query insect orthology
In addition to getting access from gene page, an ortholog group can be queried by group IDs, monarch gene IDs, or gene IDs of other species. We used the OrthoMCL algorithm to cluster protein genes of all involved species into orthology groups. Orthology page shows a list of genes that were assigned in this group and their multiple alignment results. Clickable identifiers give access to external databases.
[Query insect orthology] [Go back to the menu]
Browse biological pathways
Monarch genes have been assigned to biological pathways, according to the KEGG PATHWAY database. You can browse all pathways having monarch gene hits or select a specific pathway by inputing either a pathway ID or key words:

[Browse pathways] [Go back to the menu]
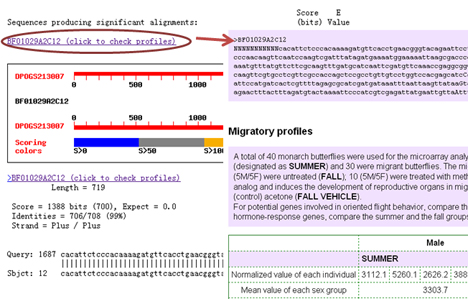
Query ESTs and migratory profiles
Migratory profiles were determined by the microarray data of brain-derived ESTs. Input an EST id to retrieve sequence and expression data directly. You can also use monarch gene ID or nucleotide sequence as inputs, and then select the appropriate hit according to the identity and location:
[Query ESTs and profiles] [Go back to the menu]
Query differentially expressed ESTs
A total of 40 monarch butterflies were used for the microarray analysis. Of the 40, 10 (5 male/5 female) were summer butterflies (designated as SUMMER) and 30 were migrant butterflies. The migrant butterflies were further divided into three groups: 10 (5M/5F) were untreated (FALL); 10 (5M/5F) were treated with methoprene (FALL METHOPRENE), which is a juvenile hormone analog and induces the development of reproductive organs in migrant butterflies; and 10 (5M/5F) were treated with vehicle (control) acetone (FALL VEHICLE). Click pull-down menu to select a pair of sampling groups for comparison. For potential genes involved in oriented flight behavior, compare the summer group to each of the three fall groups; for the juvenile hormone-response genes, compare the summer and the fall groups, and the methoprene-treated and vehicle-treated migrants. To consider sexual dimorphism, you can select considering male or female only rather than both. You also can specify the sorting method and the number of ESTs to display. Clickable identifiers give access to detailed information about ESTs.

[Query differentially expressed ESTs] [Go back to the menu]
Fetch genomic sequence
DNA sequence from v3 scaffolds can be retrieved in both the original format and reverse-complement format. This is helpful for designing primers or searching for elements on a specific segment. Please input the scaffold ID in the correct format. If the region is not designated, sequence of the entire scaffold will be returned. Please note there may be a long response time for your internet browser to load a long scaffold (our longest scaffold is >7Mb).
[Fetch genomic sequence] [Go back to the menu]
Browse expanded and contracted gene families
We analyzed lineage-specific expansion/contraction of gene families by mapping InterPro domains to OGS2.0 genes, and performed comparisons with Heliconius, Bombyx, Drosophila, and Tribolium (see phylogenetic relationship). We sort out these families according to the differences and provide a friendly browsing interface at MonarchBase. Select a family from the scroll-down menu and set a number to limit how many families listed.
[Browse expansion/contraction] [Go back to the menu]
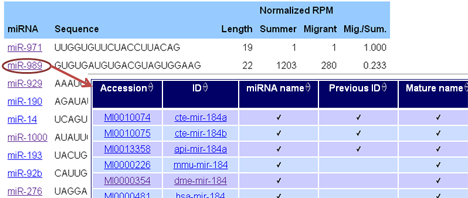
Browse microRNAs
All monarch microRNAs can be browsed with their sequences and normalized expression values. By clicking the identifiers, you can access miRBase to find homologs:
[Browse miRNAs] [Go back to the menu]
Monarch migration biology
Because monarchs are famous for their long-distance migration, the biological interpretation of the genome has focused on genes potentially involved in the migration. These genes were manually annotated and are available for browsing in catalogs, coupled with biological interpretation.
[Browse these genes] [Go back to the menu]
Other questions or suggestions?
Please contact us.